(750 × 95 px)")





INTRODUÇÃO
No Brasil, o setor de saúde tem se mantido nos últimos anos como o “2º item de gasto federal, depois de previdência, e o 3º item do gasto agregado das três esferas de governo (depois de previdência e educação)” (BRASIL, 2011). Esses gastos representam mais de 7% do Produto Interno Bruto (PIB), incluindo o gasto público (Sistema Único de Saúde – SUS) e privado, sendo que a participação privada corresponde a mais de 4% do PIB. O SUS oferece cuidados de saúde a 150 milhões de habitantes que não possuem planos privados de saúde e representa 3,9% do PIB. Esse recurso é insuficiente para atender o compromisso constitucional de acesso universal à saúde. Países com acesso universal à saúde devem investir pelo menos 8% do PIB em saúde pública. Ao contrário dos sistemas de saúde europeus, somente 44% do financiamento total dos serviços de saúde é de origem pública, aproximadamente US$ 250 públicos anuais per capita (US$ por paridade do poder de compra), o que nos coloca entre os países com menor financiamento público per capita do mundo e reforça a tese de subfinanciamento do SUS.
A promulgação da Emenda Constitucional 29 (EC29), que trata dos limites mínimos com saúde pelos municípios, estados e União, vai adicionar mais recursos financeiros para o SUS, mas ainda insuficientes para atingir os padrões de financiamento do sistema universal de saúde proposto pela Constituição de 1988.
Em contrapartida, nossa carga tributária de 39% do PIB é maior que a arrecadação fiscal dos países da OCDE (Organização para a Cooperação e Desenvolvimento Econômico), que se encontra por volta de 30% do PIB. Para agravar a situação, foram adotadas no Brasil as políticas de austeridade fiscal. Dentre as ações mais recentes adotadas para a limitação de despesas, destaca-se a Emenda Constitucional nº 95, de 2016 (EC 95/16), criando o Regime Fiscal, que estipulou um teto para a despesa primária da União e o congelamento do gasto com saúde em valores reais de 2016, por 20 anos. Tal medida reduziria, à medida que a economia crescesse, o valor do PIB do governo federal destinado à saúde bem como diminuiria o gasto per capita, desconsiderando que a população de idosos no Brasil dobrará nesse mesmo período.
No campo da epidemiologia da doença crônica, um fenômeno recente documentado em estudos observacionais brasileiros demonstra a queda na letalidade da doença cardiovascular (DCV), apesar de ainda representar a maior mortalidade (óbito por DCV na população geral), pela maior prevalência dentre as doenças crônicas não transmissíveis (DCNT). Em decorrência disso, o diagnóstico precoce e tratamento do câncer torna-se uma prioridade de saúde (Gráfico 1), já que existe maior sobrevida de pacientes com DCV somado ao aumento da longevidade da população (entre 1980 e 2009 a expectativa de vida do brasileiro experimentou um acréscimo de 10,60 anos ao passar de 62,57 anos para os atuais 73,17 anos).
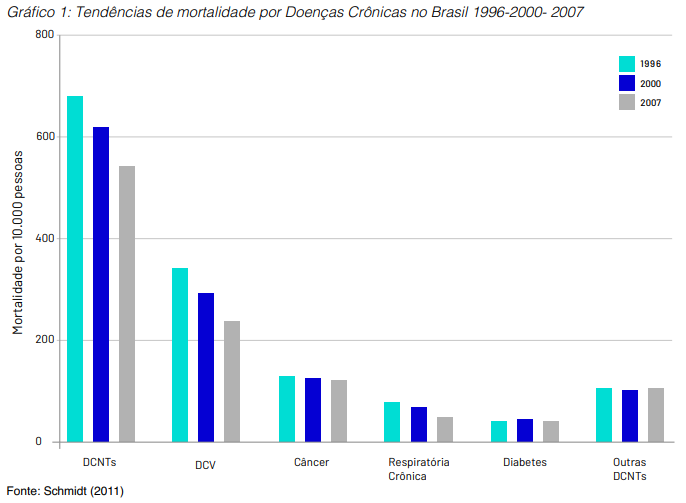
Além do aumento do número de casos, o tratamento do câncer tem maior custo unitário que a doença cardiovascular, principalmente pelo advento dos medicamentos biológicos.
O mercado global de produtos biológicos deverá atingir cerca de 291 milhões de dólares em 2020 e, em 2022, espera-se que 50% da participação do mercado farmacêutico seja em produtos biológicos.
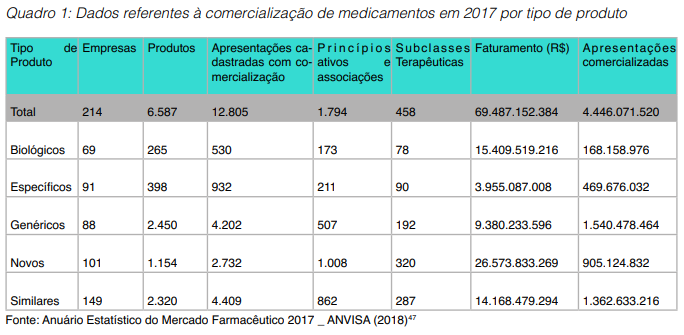
O mercado farmacêutico brasileiro movimentou em 2017 mais de 69 bilhões de reais ̶ sendo que 22% do faturamento vem de medicamentos biológicos que representaram apenas 3,8% do volume de apresentações comercializadas, como demonstrado no Quadro 1.
A Organização Mundial da Saúde (OMS) é uma agência especializada em saúde subordinada à Organização das Nações Unidas (ONU). Dentre suas várias atribuições está a publicação das diretrizes de padronização das intervenções em países membros.
Como parte de seu mandato para assegurar a qualidade global, a segurança e a eficácia dos produtos biológicos, a Organização Mundial da Saúde (OMS) forneceu normas e padrões globalmente aceitos para a avaliação destes produtos.
Produtos biológicos têm um histórico de sucesso no tratamento de muitas doenças crônicas e que trazem risco à vida. Recentemente, o vencimento de patentes e/ou proteção de dados para o primeiro grande grupo de biológicos de referência deu início a uma era de produtos que são projetados para serem “similares” a um produto original licenciado. Para seu licenciamento, esses produtos dependem, em parte, de informação prévia sobre a segurança e a eficácia obtida com os produtos de referência. A experiência clínica e o perfil de segurança estabelecido dos produtos de referência devem contribuir para o desenvolvimento de Produtos Biológicos Similares (PBs). Uma variedade de termos como “produtos biossimilares”, “produtos similares” e “produtos biológicos de entrada posterior” foram cunhados por diferentes jurisdições para descrever esses produtos.
O termo medicamento “genérico” é usado para descrever produtos medicamentosos químicos (sintéticos) de pequenas moléculas que são estrutural e terapeuticamente equivalentes a um produto original cuja patente e/ou período de proteção de dados tenha expirado. A demonstração de bioequivalência do medicamento genérico com um produto de referência é geralmente adequada e suficiente para inferir a equivalência terapêutica entre o medicamento genérico e o produto de referência. No entanto, a abordagem estabelecida para os medicamentos genéricos não é adequada para o desenvolvimento, avaliação e licenciamento dos PBS, uma vez que os produtos biológicos consistem em proteínas complexas e relativamente grandes que são difíceis de caracterizar.
O desempenho clínico dos produtos biológicos é muito influenciado pelo processo de fabricação, e alguns estudos clínicos também serão necessários para sustentar a segurança e a eficácia de um PBS. Como acontece com qualquer programa de desenvolvimento de medicamentos, o desenvolvimento de um PBS envolve uma abordagem gradual que começa com a caracterização e avaliação dos atributos de qualidade do produto, seguido por estudos não clínicos e clínicos.
Os padrões escritos estabelecidos pelo Comitê de Especialistas para a Padronização Biológica (CEPB) da Organização Mundial da Saúde servem como base para o estabelecimento dos requisitos nacionais para a produção, controle de qualidade e regulação em geral dos medicamentos biológicos. Além disso, os padrões internacionais de medida são ferramentas essenciais para o estabelecimento da potência dos medicamentos biológicos em todo o mundo. Uma gama cada vez maior de PBS está em desenvolvimento ou já está licenciada em muitos países, e a necessidade de orientações para sua avaliação e regulação em geral foi formalmente reconhecida pela OMS em 2007.
Foi reconhecido que uma série de questões importantes associadas ao uso de PB precisam ser definida pelas autoridades nacionais. Elas incluem os seguintes: questões de propriedade intelectual; intercambiabilidade e substituição do PBS com o Produto Biológico de Referência (PBR); planos de farmacovigilância e prescrição médica.
Um produto biológico de referência é utilizado como comparador para estudos de comparabilidade frente a frente com o produto biológico similar a fim de demonstrar a similaridade em termos de qualidade, segurança e eficácia. Apenas um produto original que foi licenciado com base em um dossiê de registro completo pode servir como um PBR. Não se trata de padrões de medida tais como padrões internacionais, farmacopeicos ou nacionais, ou padrões de referência.
Similaridade não é identidade. Essa afirmação resume de forma precisa a relação entre os produtos biológicos de referência, ou originadores, e os produtos biológicos similares (biossimilares), comparada à relação entre medicamentos sintéticos de referência, ou originadores, e medicamentos sintéticos genéricos.
Medicamentos sintéticos têm baixa complexidade e baixo peso molecular. Essa característica molecular permite que sejam produzidas cópias idênticas ao medicamento originador através de emprego de tecnologia farmacêutica básica e de baixo custo. Essas cópias são chamadas de medicamentos genéricos, uma vez que se trata de moléculas idênticas às dos medicamentos originadores.
Medicamentos biológicos possuem alta complexidade e alto peso molecular. Essa característica molecular aliada ao fato de que o produto biológico é cultivado a partir de um banco celular único (matriz celular), e passa por um processo de produção também único, não permite que sejam produzidas cópias idênticas ao produto originador.
Produzir uma cópia de produto biológico requer emprego de biotecnologia de alta complexidade, criação de um banco celular próprio, assim como processo de produção próprio (similar ao processo utilizado na produção do medicamento originador). Dessa forma, não existem medicamentos biológicos genéricos, tampouco “biogenéricos”. As cópias de produtos biológicos, quando comprovadas qualidade, eficácia e segurança através de estudos comparativos de fases I, II e III, serão sempre categorizadas como cópias similares aos produtos
originadores, ou “biossimilares”, pois jamais serão moléculas idênticas.
No dia 17 de dezembro de 2010, a Diretoria Colegiada da Agência Nacional de Vigilância Sanitária (ANVISA) publicou a Resolução – RDC nº 55 com objetivo de estabelecer os requisitos mínimos para o registro de “produtos biológicos novos” e “produtos biológicos” no país, para garantir a qualidade, segurança e eficácia destes medicamentos (BRASIL, 2010).
Segundo a Resolução – RDC nº 55, o “produto biológico” é o medicamento biológico não novo ou conhecido que contém molécula com atividade biológica conhecida, já registrado no Brasil e que tenha passado por todas as etapas de fabricação (formulação, envase, liofilização, rotulagem, embalagem, armazenamento, controle de qualidade e liberação do lote de produto biológico para uso). “Produto biológico novo” é o produto biológico já
registrado na ANVISA com base na submissão de um dossiê completo, e que já tenha sido comercializado no país” (originador).
A Resolução – RDC nº 55 propõe duas vias regulatórias distintas para registro dos ¨produtos biológicos¨ (biossimilares). Uma via de desenvolvimento por comparabilidade que poderá ser utilizada por um produto biológico (biossimilar) para obtenção de registro junto à autoridade regulatória, na qual foi utilizado o exercício de comparabilidade em termos de qualidade, eficácia e segurança, entre o produto desenvolvido para ser comparável e o produto “biológico novo” (originador). Outra denominada via de desenvolvimento individual “que poderá ser utilizada por um produto biológico para obtenção de registro junto à autoridade regulatória, na qual é necessária a apresentação de dados totais sobre o desenvolvimento, produção, controle de qualidade e dados não clínicos e clínicos para demonstração da qualidade, eficácia e segurança do produto, de acordo com o estabelecido nesta Resolução”.
A seguir, destacamos alguns pontos críticos desta resolução que podem comprometer a segurança, eficácia e qualidade dos produtos biológicos disponíveis no Brasil nos próximos anos.
ASPECTOS RELACIONADOS À QUALIDADE DO PRODUTO
Quanto aos critérios de qualidade do produto, a Resolução – RDC nº 55 não esclarece sobre a necessidade de estrutura molecular idêntica do produto biológico similar (PBS) em relação ao produto biológico de referência (PBR).
A diretriz da OMS é clara ao definir que a comparação da qualidade mostrando similaridade molecular entre o produto biológico similar e o produto biológico de referência é indispensável para fornecer justificativas para a previsão de que o perfil de segurança e eficácia clínica do PBR deve também se aplicar ao PBS. Pequenas diferenças no processo de fabricação podem afetar a farmacocinética, farmacodinâmica, eficácia e/ou segurança dos produtos biológicos.
De maneira ideal, o desenvolvimento de um PBS envolve a caracterização completa de um número de lotes representativos do PBR e, em seguida, a engenharia de um processo de fabricação que irá reproduzir um produto que é muito semelhante ao PBR em todos os atributos de qualidade clinicamente relevantes do produto, ou seja, os atributos do produto que podem impactar no desempenho clínico. Um PBS é geralmente derivado de um banco central de células (banco de célula mestre) separado e independente, utilizando processos de fabricação e controle próprios. Essas células devem ser selecionadas e desenvolvidas para atender aos critérios de comparabilidade necessários. Um dossiê completo de qualidade tanto para os princípios ativos quanto para os medicamentos é sempre necessário, o que atende aos padrões conforme exigidos pelas Agências Regulatórias Nacionais (ARNs) para os produtos de referência.
O amplo conhecimento sobre as relações entre as propriedades bioquímicas, físico-químicas e biológicas do produto e seus resultados clínicos facilitarão o desenvolvimento de um PBS. Para avaliar a comparabilidade, o fabricante deve realizar uma completa caracterização físico-química e biológica do PBS frente a frente com o PBR. Todos os aspectos da qualidade do produto e da heterogeneidade devem ser avaliados.
Ao realizar um exercício de comparabilidade, estudos de caracterização frente a frente são necessários para comparar o PBS e o PBR. A estrutura primária do PBS e do PBR deve ser idêntica.
NECESSIDADE DO PRODUTO BIOLÓGICO DE REFERÊNCIA PARA COMPARAÇÃO
O 1º parágrafo do Art. 27 cita que “em caso de comprovada indisponibilidade comercial do produto biológico comparador no mercado nacional e internacional, a eleição do medicamento a ser utilizado no exercício de comparabilidade deverá ser previamente discutida e anuída pela ANVISA”. Há necessidade de regras mais definidas sobre o comparador (BRASIL 2010).
Segundo a diretriz da OMS, o PBR é fundamental para o licenciamento de um PBS. As informações completas sobre o PBR fornecem a base para o estabelecimento dos perfis de segurança, qualidade e eficácia ao qual o PBS é comparado. O PBR também fornece a base para a seleção da dose e via de administração, e é utilizado em estudos de comparação exigidos para sustentar a solicitação de licenciamento. O mesmo PBR deve ser utilizado ao longo de todos os exercícios de comparabilidade (OMS, 2009).
A justificativa para a escolha do PBR deve ser fornecida pelo fabricante do PBS quando da submissão a ARN. Tradicionalmente, as ARNs têm solicitado o uso de um produto de referência licenciado nacionalmente para o licenciamento de medicamentos genéricos. O PBR deve ter sido comercializado por um período adequado e ter um volume de uso comercial de tal forma que a demonstração da similaridade a ele traz à tona um conjunto substancial de dados aceitáveis em relação à segurança e eficácia. E, mais importante, o PBR deve se licenciado com base em dados completos de qualidade, segurança e eficácia. Portanto, um PBS não deve ser considerado como uma opção ao PBR.
CARACTERÍSTICAS DOS ESTUDOS DE SEGURANÇA INCLUSIVE IMUNOGENICIDADE
Quanto às características dos estudos de segurança e imunogenicidade, a Resolução – RDC nº 55 não detalha as características necessárias do PBS para concessão do registro e não há diretrizes que orientem neste sentido. O Art. 28 da Resolução – RDC nº 55 cita genericamente que “Independentemente da via de desenvolvimento utilizada, no ato do protocolo do pedido de registro de um produto biológico novo ou produto biológico, a empresa deverá apresentar relatório do estudo de imunogeniidade” (BRASIL, 2010).
A diretriz da OMS declara que os dados de segurança pré-licenciamento devem ser obtidos em um número suficiente de pacientes para caracterizar o perfil de segurança do PBS. A comparação com o PBR deve incluir o tipo, a frequência e a gravidade dos eventos/reações adversas. Para os casos em que a eficácia similar é demonstrada em estudos confirmatórios de farmacocinética/farmacodinâmica, e os dados de segurança relevantes para a população-alvo não podem ser deduzidos desses estudos, os dados de segurança na população-alvo ainda são necessários (OMS, 2009).
Os dados de segurança devem ser comparativos. Espera-se que os dados de segurança obtidos a partir dos ensaios clínicos possam, principalmente, detectar evento/reações adversos frequentes e de curto prazo. Esses dados normalmente são suficientes para o pré-licenciamento, mas em geral é necessário um acompanhamento minucioso da segurança clínica do PBS na fase de pós-comercialização.
A imunogenicidade dos produtos biológicos deve sempre ser investigada antes do licenciamento. Mesmo que a eficácia e a segurança entre um PBS e um PBR tenham se mostrado similares, a imunogenicidade pode ainda ser diferente.
A resposta imune contra um biológico é influenciada por muitos fatores, tais como a natureza do princípio ativo, impurezas relacionadas ao produto e ao processo, excipientes e estabilidade do produto, via de administração, regime de dose, além de fatores relacionados ao paciente, à doença e a terapia. As consequências da imunogenicidade indesejada podem variar consideravelmente, desde clinicamente irrelevante até grave e potencialmente fatal.
INTERCAMBIABILIDADE
A intercambiabilidade é uma propriedade molecular que, em teoria, confere aos medicamentos a possibilidade de que sejam intercambiáveis sem que haja prejuízo para o paciente na jornada de seu tratamento. Essa atividade é bastante comum em terapias com produtos sintéticos, promovendo trocas entre medicamentos originadores e genéricos.
Devido ao fato de medicamentos biológicos não serem idênticos e levando em conta seu alto potencial imunogênico, o intercâmbio entre produtos desta categoria requer discussões objetivas e muita cautela por parte dos atores desse processo: agente regulador, paciente, médico assistente, agente financiador da terapia (público e privado), indústria farmacêutica.
Entendemos que a intercambiabilidade deve ser definida pela autoridade reguladora ou de saúde. Tomando como exemplo algumas agências de referência internacional, o FDA (Food and Drug Administration – EUA) define que um medicamento biossimilar pode ser considerado intercambiável se:
• Seu resultado clínico esperado for igual àquele produzido por um produtor de referência em qualquer paciente;
• A alternância ou troca repetida entre esses medicamentos não apresente risco adicional à segurança ou redução de eficácia na comparação com o uso continuado do produto de referência.
Em 2019, o FDA publicou o documento Considerations in Demonstrating Interchangeability With a Reference Product Guidance for Industry, que determina os protocolos de estudos a serem desenvolvidos para comprovar eficácia e segurança em possíveis intercâmbios envolvendo medicamentos biológicos originadores e seus biossimilares.
No caso da agência europeia de medicamentos (EMA), a avaliação não inclui recomendações sobre a possibilidade de utilizar um biossimilar de forma intercambiável com seu medicamento de referência, o dossiê não contém evidências que fundamentem uma determinação sobre a intercambiabilidade, além de determinar que “para perguntas relacionadas à troca de um medicamento biológico por outro, o paciente deve conversar com seu médico e farmacêutico”. A decisão legal sobre a intercambiabilidade cabe aos Estados Membros (EMA, 2012).
Apesar da definição de intercambiabilidade proposta pelo FDA, não há um consenso mundial quanto ao melhor desenho de estudos clínicos que possa ser usado para confirmar e evidenciar este conceito. É possível até que o desenho supostamente ideal seja inexequível pelo seu tamanho e custo.
Em 2012, Ebbers, Muenzberg e Schellekens realizaram um estudo de revisão de literatura chamado “The safety of switching between therapeutic proteins” (“A segurança na alternância entre proteínas terapêuticas”). Nessa revisão, fornecem uma visão geral de dados relacionados à alternância entre eritropoietina, hormônio de crescimento e (G-CSF) fator estimulador de colônias de granulócitos. O estudo abrange tanto a alternância (switch) entre produtos inovadores dentro da mesma classe quanto a de inovadores para biossimilares e vice-versa.
Os dados sobre a frequência de alternância na prática clínica são escassos, mas os autores não encontraram nenhuma evidência a partir desses limitados dados de ensaios clínicos ou de vigilância pós-comercialização que indicassem que essa alternância de diferentes produtos biofarmacêuticos levasse a preocupações de segurança. É verdade também que a maioria dos estudos revisados não foi desenhada para identificar efeitos adversos relacionados à alternância.
Alguns estudos somente acompanharam pacientes depois da alternância num estudo “open label” (estudo aberto) e de um único braço, ou seja, estudos em que todas as partes envolvidas (paciente, médico e coordenador) são informadas sobre qual droga está sendo utilizada, assim como a dose, por cada um dos participantes. Em estudo aberto, não se utiliza placebo, ou seja, temos apenas um grupo de pacientes observado (único braço). Essa revisão de literatura, apesar de bem realizada, não nos dá robusta evidência que sustente que a intercambiabilidade entre esses produtos, dentro de suas classes terapêuticas, é segura. Além disso, trata-se de medicamentos biológicos bem menos complexos do que anticorpos monoclonais ou proteínas de fusão, por exemplo.
Também há incertezas quanto à população que deve ser incluída nesses estudos (BPCI, 2010; EBBERS et al., 2012):
1. Para doenças crônicas, deve-se estudar tanto novos pacientes quanto pacientes estáveis que já receberam tratamento?
2. Estudos sobre a alternância devem ser conduzidos com as populações mais sensíveis?
3. Subpopulações com diferentes perfis de eficácia ou segurança (p.ex., pediátricas)
4. Outros possíveis fatores para consideração:
• Risco de imunogenicidade (frequência, severidade, anticorpos antidrogas);
• Segurança e eficácia em diferentes indicações;
• Indicações extrapoladas.
A ANVISA, através de sua nota de esclarecimento publicada em 2017 e revisada em outubro de 2018, cujo objeto é intercambiabilidade e substituição de produtos registrados pela via de desenvolvimento por comparabilidade (“biossimilares”) e o produto biológico comparador (originador), afirma: “Ressalta-se que a ANVISA, em consonância com a atuação de agências reguladoras de outros países, não classificará os produtos biossimilares como intercambiáveis ou não” (ANVISA, 2018, p.1).
Ainda no mesmo documento, a ANVISA afirma:
Importante ressaltar que a avaliação médica e a adequada atenção farmacêutica são imprescindíveis no caso de trocas de produtos biossimilares e seus comparadores, tanto para fins de prescrição e uso adequado do produto quanto para fins de farmacovigilância e acompanhamento pós-mercado desses produtos. A GPBIO também entende não serem adequadas múltiplas trocas entre produtos biossimilares e o produto biológico comparador, ficando a rastreabilidade e monitoramento do uso bastante dificultados nestes casos (ANVISA, 2018, p.1).
IMPACTO NA FARMACOVIGILÂNCIA
A vigilância pós-comercialização de uma nova e complexa classe emergente de anticorpos biossimilares é crítica.
A substituição pode comprometer a eficácia da farmacovigilância:
• Se os médicos não estiverem informados, sua capacidade de atribuir eventos adversos ao agente apropriado pode ser prejudicada.
• Se a reação adversa tiver início tardio: algumas reações adversas – incluindo reações imunogênicas – podem se desenvolver somente após vários meses de tratamento.
• Outras situações podem ser bem desafiantes:
• O médico que atende o paciente portador de um efeito adverso, pode não ser o médico que prescreveu o medicamento biológico.
• A caixa do remédio pode ter sido descartada pelo paciente.
• Falta de clareza ou acurácia por parte do paciente na hora de reportar o efeito adversos.
• A adoção de uma nomenclatura distinguível para cada medicamento biológico, como um “sobrenome”, poderia facilitar o monitoramento e a rastreabilidade do produto e do paciente, no caso de haver efeitos adversos.
O FDA publicou em agosto de 2015 seu guia específico para nomenclatura de medicamentos, sugerindo que seja adicionado um sufixo de 4 letras ao nome do Ingrediente Farmacêutico Ativo (IFA) de cada medicamento aprovado.
CENÁRIO BRASILEIRO DE UTILIZAÇÃO DE MEDICAMENTOS BIOLÓGICOS ORIGINADORES E BIOSSIMILARES
Uma prática cada vez mais comum em nosso país, e bastante controversa, é a alternância de agentes biológicos em pacientes com adequada e bem tolerada terapia, que denominamos troca não médica.
A motivação por trás desta alternância capitaneada pelas fontes pagadoras da terapia é geralmente a potencial redução de custos22,23, que muitas vezes não se confirmará diante do risco de uma provável intolerância imunológica do paciente e consequente perda desta linha terapêutica.
Por isso, a comunidade científica trata o tema com muito cuidado e precaução, uma vez que em teoria é real a possibilidade de surgirem, no médio e no longo prazo, reações relacionadas à imunogenicidade, como a criação de anticorpos antidroga, principalmente quando se tratar de pacientes com tratamento em curso e com resultado satisfatório. Nesse caso, a alternância poderia representar um risco indesejado.
Não existem no momento evidências robustas que suportem a intercambiabilidade, principalmente levando-se em consideração que a alternância poderá ser não apenas entre o medicamento inovador e um biossimilar, mas entre o inovador e todos os seus biossimilares aprovados e disponíveis no mercado.
Existem hoje no Brasil moléculas com mais de um medicamento biossimilar correspondente aprovado. Além das múltiplas trocas que podemos assistir entre inovadores e biossimilares, uma possível troca entre estes medicamentos biossimilares (biossimilar x biossimilar) nos traz uma preocupação ainda maior, um acúmulo de incertezas, uma vez que nos estudos comparativos para registro destes medicamentos, todos os biossimilares são comparados a um único medicamento de referência. Portanto, não existe um estudo comparativo biossimilar x biossimilar.
Sequer temos evidências de comprovação de biossimilaridade entre dois medicamentos biossimilares. Na prática, estaremos promovendo o intercâmbio entre duas moléculas cujo grau de similaridade nem é conhecido. Nesse cenário, aumentamos ainda mais o alto risco relacionado à intercambiabilidade.
Assim como não temos regulamentação específica para intercambiabilidade em nenhum país da América Latina, tampouco encontramos nas normas regulatórias de farmacovigilância na região que determine diferenciação de nomenclatura entre o medicamento inovador e seu biossimilar.
No Brasil, além do mecanismo de compras públicas por licitação, que aumenta o risco de substituição automática entre medicamentos biológicos inovadores e biossimilares, as prescrições não po-dem ter nomes de marcas quando prescritas no cenário do SUS, causando ainda mais impacto na rastreabilidade.
GRUPO DE TRABALHO OBJETO DA PORTARIA GM/MS Nº 1.160, DE 3 DE MAIO DE 2018 – DIRETRIZES PARA UMA POLÍTICA NACIONAL DE MEDICAMENTOS BIOLÓGICOS NO ÂMBITO DO SUS
O Grupo de Trabalho (GT) para a Política de Medicamentos Biológicos foi instituído pela Portaria GM/MS nº1.160, no dia 3 de maio de 2018 e realizou durante o segundo semestre de 2018 reuniões deliberativas para compor as diretrizes para o estabelecimento desta política. O Conselho Nacional de Saúde, esteve representado neste GT pelas conselheiras nacionais de saúde Ana Lúcia Marçal, da Associação Brasileira Superando o Lúpus, e Eliane Cunha, do Sindicato dos Servidores do Sistema Nacional de Auditoria do SUS (UNASUS).
A expectativa do grupo era que a Política Nacional de Medicamentos Biológicos no âmbito do SUS entrasse em vigor no início de 2019, levando em consideração as contribuições do controle social na saúde. No entanto, em setembro de 2019, o Ministério da Saúde publicou o resultado das discussões do GT por meio de um relatório composto por 211 páginas, determinando que esta foi a base para a discussão e formulação da Política Nacional
de Medicamentos Biológicos no âmbito do Sistema Único de Saúde.

O resultado deste GT não levou em consideração as contribuições do controle social e, até o momento, não foi fomentada a criação de uma política nacional de medicamentos biológicos no âmbito do SUS. Considerando uma ampla análise deste relatório, a Biored Brasil manifestou-se por meio de um posicionamento público, solicitando urgente atenção das autoridades e, principalmente, do Ministério da Saúde nos seguintes itens:
• Aspectos técnicos e de segurança do paciente são primordiais e nunca podem ser ignorados para se cumprir contratos comerciais com empresas.
• Pedimos a suspensão imediata da Nota Técnica 655/2019, o abastecimento das farmácias públicas com ambos os medicamentos em questão. Junto a isso, uma orientação clara de que os medicamentos não são intercambiáveis e que o uso do biossimilar é autorizado somente para pacientes naïve (que nunca utilizaram etanercepte) ou para pacientes em que o médico expressamente autorizou sua troca. Já os pacientes em uso do biossimilar, também não podem ser trocados ao originador sem a anuência expressa em relatório médico.
• Criação de mecanismo de identificação único para cada medicamento a ser acrescido ao INN (DCB), que diferencie os medicamentos de referência e todos os seus biossimilares, ou a adoção de um mecanismo universal de identificação única como o “Qualificador Biológico” em discussão pela Organização Mundial de Saúde e já adotado pela FDA. Ou ainda, o uso da marca/fabricante no SUS, conforme adotaRepresentantes do Conselho Nacional de Saúde Ana Lúcia Marçal e Eliane Cunha do pela União Europeia e já recomendado pela ANVISA.
• As múltiplas trocas devem ser desencorajadas aos médicos e o sistema de dispensação deve coibir que aconteça qualquer troca de forma fortuita, ao utilizar um sistema robusto de controle e dispensação.
• Criação de documento a ser entregue ao paciente no momento da entrega e/ou administração do medicamento (Cartão do Paciente em uso de biológico), contendo informações como nome do paciente, nome do médico assistente, nome comercial, fabricante, lote, país de origem, prazo de validade, INN (DCB) com qualificador biológico. A Biored criou em 2018 o BIOCARD, projeto já com piloto em andamento e que estamos à disposição do Ministério da Saúde para apresentar proposta [25].
• Criação de políticas públicas sobre intercambiabilidade, com a participação ativa das sociedades científicas e sociedade civil organizada (organização de pacientes).
• Promoção de políticas no SUS com soluções práticas que garantam a segurança dos pacientes no uso de medicamentos biológicos, com boas práticas de aquisição, distribuição, dispensação, administração e farmacovigilância.
• Pedimos uma revisão da política pública de PDPs de biológicos, que coloca o paciente brasileiro em risco de múltiplas trocas, por ter vários fabricantes reproduzindo cópias de uma mesma molécula e ainda inibem uma economia ao fixar preços.
• Transparência e participação pública, principalmente dos pacientes e suas organizações, bem como da sociedade médica científica, nas decisões que afetem os tratamentos. (BIORED, 2019).
CONCLUSÃO
O desafio de oferecer assistência a populações mais longevas, com grande carga de doenças crônico-degenerativas, é maior diante da pesquisa e desenvolvimento de novas tecnologias, principalmente de origem biotecnológica, que melhoram desfechos de morbi-mortalidade e qualidade de vida, mas têm grande impacto orçamentário. Outro ponto fundamental desta discussão é a qualidade dos produtos, pois o caminho fácil de aumentar acesso sem estabelecer critérios rígidos de qualidade é preocupante, principalmente no tópico dos medicamentos de origem biológica. Os danos à saúde podem superar os benefícios, caso as recomendações da diretriz OMS não sejam adotadas.
Os medicamentos biossimilares de anticorpos monoclonais têm um histórico de utilização ainda muito limitado, o que dificulta uma boa avaliação pós-comercialização pelo pouco tempo de mercado. Até o momento não existem evidências robustas de que, no caso dos anticorpos monoclonais, a intercambiabilidade entre produtos biológicos e seus biossimilares acarretará problemas clínicos relevantes. No entanto, tampouco temos evidências que suportem essa intercambiabilidade, principalmente se levarmos em consideração que a alternância poderá ser não apenas entre o medicamento inovador e um biossimilar, mas entre o inovador e todos os seus biossimilares aprovados e disponíveis no mercado, além de trocas entre biossimilares que sequer foram alvo de estudos de similaridade. Por isso, o tema deve ser tratado com muito cuidado e precaução.
A intercambiabilidade entre medicamentos biotecnológicos originadores e seus biossimilares já é uma prática no Brasil, principalmente no setor público, o maior comprador, uma vez que suas compras são realizadas na modalidade licitatória. Sendo assim, defendemos que ações sejam realizadas para minimizar as consequências dessa situação, como revisões no processo licitatório e um controle rigoroso na dispensação dos medicamentos e na rastreabilidade de efeitos adversos, com a adoção de identificação individualizada.
Faz-se imprescindível a adoção, por parte das empresas farmacêuticas envolvidas, de programas de gestão e minimização de riscos que promovam, dentre outras coisas, educação de profissionais de saúde e pacientes, com a devida auditoria da autoridade regulatória.
São grandes os desafios para a farmacovigilância dos medicamentos biológicos e biossimilares devido à sua complexidade e pequenas diferenças estruturais que podem acarretar diferentes reações imunológicas, principalmente em casos de alternância na utilização desses medicamentos.
O sistema de saúde precisa estar preparado estruturalmente para essa realidade, apoiado em programas educacionais e regulamentações robustas que possam facilitar a rastreabilidade dos medicamentos biológicos.
Acreditamos que a comunidade científica e acadêmica, além das sociedades civis organizadas (pacientes), devem ser aliados do Ministério de Saúde e da Anvisa na discussão de possíveis regulamentações, na disseminação de conhecimento e em atividades de farmacovigilância ativa.
Este artigo faz parte do livro “Ciência, Tecnologia, Vigilância em Saúde e Assistência Farmacêutica, políticas públicas oriundas do controle social, garantidoras de democracia, soberania nacional e acesso à saúde”.
Baixe a cópia gratuita do livro em https://editora.redeunida.org.br/wp-content/uploads/2022/01/Livro-Ciencia-Tecnologia-Vigilancia-em-Saude-e-Assistencia-Farmaceutica-politicas-publicas-oriundas-do-controle-social-garantidoras-de-democracia-soberania-nacional-e-acesso-a-saude.pdf
Livro-Ciencia-Tecnologia-Vigilancia-em-Saude-e-Assistencia-Farmaceutica-politicas-publicas-oriundas-do-controle-social-garantidoras-de-democracia-soberania-nacional-e-acesso-a-saude
Descubra mais sobre Artrite Reumatóide
Assine para receber nossas notícias mais recentes por e-mail.