(750 × 95 px)")






Como são desenvolvidos os medicamentos biossimilares?
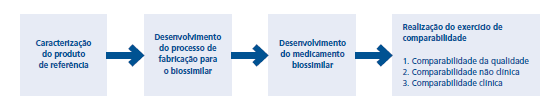
Os medicamentos biossimilares são desenvolvidos por meio de uma abordagem sistemática e passo a passo. O objetivo dessa abordagem é garantir que um medicamento biossimilar seja altamente semelhante ao produto de referência em termos de qualidade, segurança e eficácia. Primeiro o produto de referência é definido utilizando-se ferramentas sofisticadas de análise e o conhecimento clínico existente. Emprega-se um processo de desenvolvimento e fabricação para o medicamento biossimilar buscando igualar a variabilidade do produto de referência. Depois que o produto é fabricado, ele é comparado com o de referência em um exercício de comparabilidade biossimilar.
Há três passos no exercício de comparabilidade
O primeiro busca mostrar que a qualidade é comparável. Nesse estágio, as qualidades físico-químicas e biológicas são comparadas por meio de uma série de testes analíticos.
O segundo passo é a comparabilidade não clínica. Isso inclui estudos de dosagem e a análise do que o organismo faz com o medicamento e o que este faz com o organismo em modelos animais adequados. O objetivo é detectar qualquer diferença entre o medicamento biossimilar e o produto de referência.
O terceiro passo é a comparabilidade clínica, no qual o medicamento biossimilar é testado em seres humanos em um ensaio clínico. Nesse estágio, também deve ser mostrado um perfil de segurança comparável em termos de gravidade e frequência de efeitos colaterais.
As comparabilidades não clínica e clínica dão a segurança de que qualquer diferença observada na esfera da comparabilidade de qualidade não terá impacto sobre a segurança e eficácia do medicamento biossimilar. Os dados clínicos não têm como objetivo mostrar os benefícios do medicamento, e sim garantir que qualquer diferença não terá impacto sobre sua qualidade e segurança. A quantidade de dados não clínicos e clínicos necessários depende do quanto esses dados são completos e do produto ou tipo de produto. Muitas vezes os estudos clínicos para medicamentos biossimilares são menores e mais curtos do que aqueles sobre os produtos de referência.
Depois que todos os dados do exercício de comparabilidade tiverem sido coletados, são apresentados ao órgão regulador competente, junto com um plano de gestão de risco. Esse plano descreve o perfil de segurança do medicamento e esboça como o fabricante vai monitorar em mais detalhes e preencher qualquer lacuna nos dados com relação à segurança e eficácia. O órgão regulador avaliará os dados de comparabilidade, o plano de gestão de risco e os planos de monitoramento depois que o medicamento for disponibilizado para decidir se o biossimilar deve ser aprovado.
Como são regulamentados os biossimilares e quem os regulamenta?
Os medicamentos biossimilares precisam de uma via regulatória diferente da via de um novo medicamento biológico ou de um genérico. A via é muitas vezes chamada de uma via abreviada de licenciamento, já que é uma versão mais curta da via utilizada para aprovar um medicamento biológico novo. União Europeia abriu o caminho para a regulamentação e aprovação dos medicamentos biossimilares. Desde o Relatório informativo da IAPO de 2006 houve vários avanços em sua regulamentação mundialmente.
Principais perguntas a serem respondidas pelos reguladores ao considerarem a aprovação de um medicamento biossimilar:
- Foram fornecidos os dados de todos os passos do exercício de comparabilidade (qualidade, não clínicos, clínicos)?
- Foram realizados testes para possíveis reações imunes?
- Foi elaborado um plano para gestão de risco após a aprovação?
- Há um plano de farmacovigilância em vigor para detectar efeitos adversos?
Fonte: IAPO Américas

Descubra mais sobre Artrite Reumatóide
Assine para receber nossas notícias mais recentes por e-mail.